MAJIQ-L
MAJIQ-L takes an input three sources of information: Transcriptome annotation; short reads processed by MAJIQ v2; and long reads in gtf format, processed by the user’s algorithm of choice. It then computes and displays an extensive set of statistics that contrast the available annotation and the two sequencing sources in terms of novel junctions, introns, coverage, inclusion levels, etc. such that existing gaps between the three sources can be captured
Using the three input sources, MAJIQ-L constructs unified gene splice graphs with all isoforms and all LSVs visible for analysis. This unified view is implemented in a new visualization package (VOILA v3), allowing users to inspect each gene of interest where the three sources agree or differ.
Note: for additional scripts/software required to generate all figures from the MAJIQ-L paper, including general purpose algorithms, please see the supplementary MAJIQ-L code repository here
VOILA lr: Unified Visualization
VOILA lr takes four inputs:
A splicegraph database (splicegraph.sql from MAJIQ Builder)
Voila files (SAMPLE_ID.psi.voila from MAJIQ quantifiers)
Long reads in gtf format, processed by the user’s algorithm of choice
A SAMPLE_ID.tsv, a tab separated file, containing transcript read number processed by the user’s algorithm of choice like:
transcript_id_1 transcript_read_1transcript_id_2 transcript_read_2transcript_id_3 transcript_read_3
For example, use the following command options:
voila lr
–lr-gtf-file /PAHT/TO/SAMPLE_ID.gtf
–lr-tsv-file /PAHT/TO/SAMPLE_ID.tsv
-sg /PATH/TO/splicegraph.sql
-o OUTPUT_FOLDER
Default: 1 |
Output is the resulting long read voila file (recommended extension in .lr.voila format)
To display the unified splicegraph, the output of voila lr (SAMPLE_ID.lr.voila) is given as input to VOILA v3. Users may use voila view as a server to display results as shown here. For example, use the following command options:
voila view
/PATH/TO/splicegraph.sql
/PAHT/TO/SAMPLE_ID.psi.voila
/PATH/TO/SAMPLE_ID.lr.voila
-p 7050
–host 0.0.0.0
Default: 1 |
Additional details on usage can be found by adding –help to the subcommand of interest (e.g. voila view –help)
Examples of obtaining GTF and TSV files from different LR algorithms
Using IsoQuant:
GTF file: Obtain the GTF file containing the discovered expressed transcripts (
SAMPLE_ID.transcript_models.gtfshould be provided) by running IsoQuant.TSV file: Obtain the TSV file with read counts assigned to each transcript (
SAMPLE_ID.transcript_model_counts.tsvshould be provided) by running IsoQuant.
Using FLAIR:
GTF file: Obtain the GTF file containing the discovered expressed transcripts from the flair collapse step.
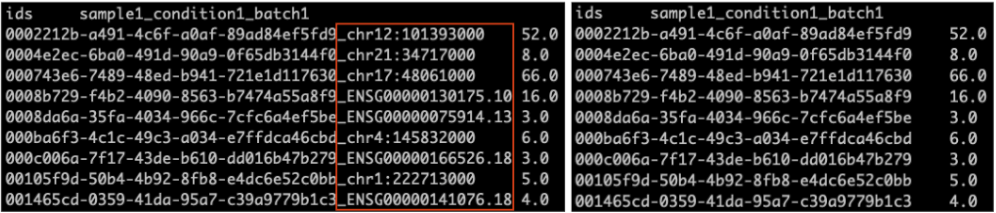
TSV file: Obtain the TSV file with read counts assigned to each transcript from the flair quantify step. The output of
flair quantifylooks like the screenshot on the left below. You need to modify this file by removing the gene_id after the last underscore in the transcript IDs to match the format shown in the screenshot on the right. This modified TSV file should be provided as your input TSV file.
VOILA lr
- usage: voila lr [-h] [–voila-file VOILA_FILE] [–gene-id GENE_ID]
[–only-update-psi] [–lr-gtf-file LR_GTF_FILE] [–lr-tsv-file LR_TSV_FILE] -o OUTPUT_FILE [-sg SPLICE_GRAPH_FILE] [-j NPROC] [–debug] [–memory-map-hdf5]
- optional arguments:
- -h, --help
show this help message and exit
- --voila-file VOILA_FILE
This should be a .psi.voila file which we will match LSV definitions to to run the beta prior. If not provided, PSI values will not be rendered for long read LSVs
- --gene-id GENE_ID
Limit to a gene-id for testing
- --only-update-psi
Instead of re generating all data, only update the PSI values. Requires -o to point to an existing .lr.voila file, and –voila-file to be provided as well
- -j NPROC, --nproc NPROC
Number of processes used to produce output. Default is half of system processes.
- --debug
Show Verbose output
- --memory-map-hdf5
by default, hdf5 voila files will be opened and read as needed, however, for greater performance it may help to instead preload these files into memory, if your server has sufficient RAM. Use this option to memory map the files. If used with view mode, you must also specify an index file to save to with –index-file
- -l LOGGER, --logger LOGGER
Set log file and location. There will be no log file if not set.
- --silent
Do not write logs to standard out.
- required named arguments:
- --lr-gtf-file LR_GTF_FILE
path to the long read GTF file
- --lr-tsv-file LR_TSV_FILE
path to the long read TSV file
- -o OUTPUT_FILE, --output-file OUTPUT_FILE
the path to write the resulting voila file to (recommended extension .lr.voila)
- -sg SPLICE_GRAPH_FILE, –splice-graph-file SPLICE_GRAPH_FILE
the path to the majiq splice graph file which will be used to align to annotated exons
Citation
The paper describing MAJIQ-L algorithm is available at https://www.biorxiv.org/content/10.1101/2023.11.21.568046v1.