Quick Start
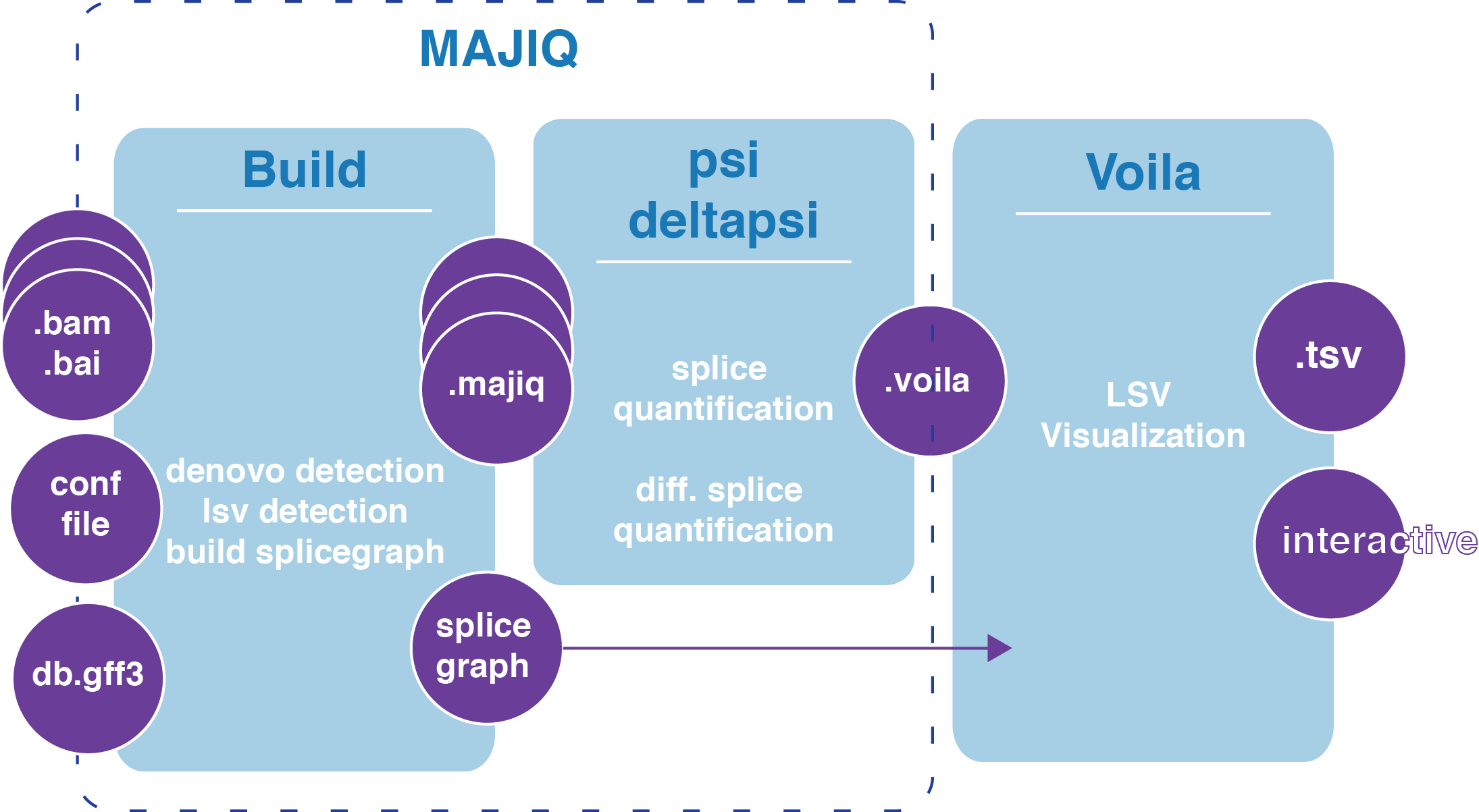
MAJIQ tool is based in three main steps, MAJIQ builder, MAJIQ quantification and VOILA, Each one of them has an specific function added to it, detect and annotate the splicegraph and events, quantify those events, and visualize the output.
Before MAJIQ
MAJIQ requires three types of files, sequence files in bam format (including the index in bai format), the annotation DB in gff3 format or (DB.gff3), and the configuration file (or conf file).
Select a GFF3 annotation file
The general feature format (gene-finding format, generic feature format, GFF) is a file format used for describing genes and other features of DNA, RNA, and protein sequences. The format specification for the gff version 3 can be found at GFF3 format. In our case we use some of these features in order to define genes, transcripts and exons. An example of this format is shown below
- chr1 protein_coding gene 107399655 107452689 . + . Name=Serpinb7;ID=ENSMUSG00000067001;Name=ENSMUSG00000067001
- chr1 protein_coding mRNA 107399655 107435399 . + . Parent=ENSMUSG00000067001;Name=Serpinb7-002;ID=ENSMUST00000154538
- chr1 protein_coding exon 107399655 107399724 . + . Parent=ENSMUST00000154538;ID=exon:ENSMUST00000154538:1
- chr1 protein_coding exon 107428231 107428416 . + . Parent=ENSMUST00000154538;ID=exon:ENSMUST00000154538:2
- chr1 protein_coding exon 107434736 107434786 . + . Parent=ENSMUST00000154538;ID=exon:ENSMUST00000154538:3
- chr1 protein_coding exon 107435327 107435399 . + . ID=exon:ENSMUST00000154538:4;Parent=ENSMUST00000154538
- chr1 protein_coding five_prime_UTR 107399655 107399724 . + . Parent=ENSMUST00000154538;ID=five_prime_UTR:ENSMUST00000154538:1
- chr1 protein_coding five_prime_UTR 107428231 107428248 . + . ID=five_prime_UTR:ENSMUST00000154538:2;Parent=ENSMUST00000154538
- chr1 protein_coding start_codon 107428249 107428251 . + 0 Parent=ENSMUST00000154538;ID=start_codon:ENSMUST00000154538:1
- chr1 protein_coding CDS 107428249 107428416 . + 0 ID=CDS:ENSMUST00000154538:1;Parent=ENSMUST00000154538
- chr1 protein_coding CDS 107434736 107434786 . + 0 Parent=ENSMUST00000154538;ID=CDS:ENSMUST00000154538:2
- ...
It is important to note that MAJIQ makes some assumptions when parsing the hierarchical GFF3 file and currently has some specific requirements:
- We only consider sequence features with the type (column 3) “gene”
- For every gene, we only consider isoforms of a gene with a type of “mRNA” or “transcript”
- All entries (except for genes) should have a parent attribute
- All genes should have a unique ID attribute
- Within a gene all entries should have a unique ID attribute.
- A gene can have a Name attribute, otherwise the ID will be used instead in the output.
Keeping these in mind will be important for analyzing the types of transcripts you care about and modifying your GFF3 annotation file may be necessary.
In order to obtain this format, we recommend the use of some of the most well known online DB. They provide the annotation files in some format like GTF, and you can transform this file to GFF3 using a script, like this script
You can also download the annotation files used in Vaquero-Garcia et al., 2016 for the Ensembl hg19 or mm10 genome builds here.
Study configuration file
MAJIQ has a set of parameters needed for its execution. Several of them depend of the RNA-Seq study. This configuration file should include this information in order to be able to pass it the the MAJIQ Builder. Secondly, it is useful to keep the info of the study ready and accessible.This is an example of the configuration file, divided in two blocks, info and experiments:
- [info]
- readlen=76
- bamdirs=/data/MGP/ERP000591/bam[,/data/MGP2/ERP000591/bam]
- genome=mm10
- strandness=none[|forward|reverse]
- [experiments]
- Hippocampus=Hippocampus1,Hippocampus2
- Liver=Liver1,Liver2
- [optional]
- Hippocampus1=strandness:none
- Liver2=strandness:reverse
Info This is the study global information needed for the analsysis. The mandatory fields are:
- readlen: Length of the RNA-seq reads. MAJIQ can handle experiments with multiple read lengths, just indicating the longest read length
- bamdirs: Comma separated list of paths where the bam files are located. If two files with the same names are in different paths, the first usable path is chosen.
- genome: Genome assembly
- strandness=forward[|reverse|none]: Some of the RNASeq preparations are strand specific. This preparations can be reverse strand specific[reverse], forward strand specific [forward], or non-strand specific [none]. This parameter is optional, in which case None is used as default.
Experiments This section defines the experiments and replicates that are to be analyzed. Each line defines a condition and its name can be customized in the following way:
<group name>=<experiment file1>[,<experiment file2>]
where the experiment file is the sorted bam filename inside the samdir directory (excluding extension
.bam). MAJIQ expects to find within the same directory bam index files for each experiment with
the
format <experiment file>.bam.bai.
Multiple replicates within an experiment should be comma separated with no spaces.
Optional
This section allows the user to specify changes in the global parameters specific to single experiments.
The
syntax goes as follows,
<experiment file1>=<option1>:<value1>,...,<optionN>:<valueN>
The user only need to add the experiments that have different parameter value than the global parameters, using this section only when is needed.
Currently only strandness has been adapted to be overwrite using this optional section, but new options can be added in the future.
MAJIQ Builder
MAJIQ Builder is the part of MAJIQ tool where RNA-Seq data is analyzed in order to detect LSV candidates.
All conditions and replicates that will be analyzed with MAJIQ PSI or delta PSI should be executed TOGETHER in a single Builder execution.
majiq build <transcript list> -c <configuration file> -j NT -o <build
outdir>
Transcriptome annotation: This is the file with the annotation database. Currently, we accept only GFF3 format. For a better description, see the annotation file section.Configuration file: This is the configuration file for the study. This file should define the files and the paths for the bam files, the read length, the genome version, and some other information needed for the builder. For a more detailed information, please check the configuration file section.NT: Number of threads to use.Build outdir: Directory where the output will be placed. MAJIQ builder has a set of output files including one .majiq for each bam file and one splicegraph.sql. These files will be the input files in the next steps of the analysis.
MAJIQ Builder has several arguments in order to tweak its analysis and performance. Please check the MAJIQ parameters section for a more detailed explanation.
PSI Analysis
PSI quantification
MAJIQ PSI quantifies the LSV candidates given by the Builder. In order to improve its accuracy and reproducibility, it allows the use of biological replicates.majiq psi <build outdir>/<replicate1>.majiq [<build outdir>/<replicate2>.majiq
...] -j NT -o <psi outdir> -n <cond_id>
*.majiq file[s]: the path to the .majiq file(s) that were created by the MAJIQ Builder execution.cond_id: group identifier that you want to use for this executionNT: Number of threads to use.Please check the MAJIQ parameters section for a more detailed explanation of all the arguments.
Delta PSI quantification
Majiq Delta PSI quantifies the differential splicing between two different groups (or conditions). Like PSI, Delta PSI is able to use replicates for each group in order to improve its accuracy and reproducibility.
majiq deltapsi -grp1 <build outdir>/<cond1_rep1>.majiq [<build outdir>/<cond1_rep2>.majiq
...] -grp2 <build outdir>/<cond2_rep1>.majiq [<build outdir>/<cond2_rep2>.majiq
...]
-j NT -o <dpsi outdir> -n <cond1_id> <cond2_id>
-grp1 .majiq file[s]: Set of .majiq file[s] for the first condition,-grp2 .majiq file[s]: Set of .majiq file[s] for the second condition,--name cond_id1 cond_id2: group identifiers for grp1 and grp2, respectively, used for naming output filesNT: Number of threads to use.
Please check the MAJIQ parameters section for a more detailed explanation of all the arguments.
Visualize results with VOILA
There are two ways too visualize PSI/Deltapsi quantification with Voila. We can generate a text tsv (tab separated values) file or we can use an interactive visualization tool.By default VOILA uses a threshold of a change of |dPSI| >= 0.2 (20%) between conditions. To change
this
threshold you can use the option --threshold and specify a fraction from 0
to 1.
To show all
LSVs --show-all can be used instead.
Human-readable text output file
In order to use MAJIQ/VOILA output as an input for post-quantification analysis you can generate VOILA tsv file. This file is a tab separated value file where each line is a single LSV. As an example, some of the columns are gene id, lsv id, Expected PSI or DeltaPSI, confidence or junction quantification, and many others. For a more comprehensive VOILA tsv description please refer to VOILA section. The tsv file can be generated executing
voila tsv <build outdir>/splicegraph.sql <dpsi outdir>/<cond1_id>_<cond2_id>.deltapsi.voila
-f output_file
<dpsi_outdir>/<cond1_id>_<cond2_id>.deltapsi.voilais the output file from delta PSI computation,splicegraph.sqlcontains information about the genes and splice variants identified in the MAJIQ Builderoutput_fileis the tsv output file name
Interactive VOILA
The interactive version of VOILA requires a web browser from wich you can visualize the local web application that VOILA spawns. To visualize PSI/DeltaPSI quantification with an interactive Voila tool execute:
voila view <build outdir>/splicegraph.sql <dpsi outdir>/<cond1_id>_<cond2_id>.deltapsi.voila
<dpsi_outdir>/<cond1_id>_<cond2_id>.deltapsi.voilais the output file from delta PSI computation,splicegraph.sqlcontains information about the genes and splice variants identified in the MAJIQ Builder
In the output directory <voila outdir> you will find:
index.html: HTML file with a table containing all genes and LSVs identified and analyzed and links to more detailed summaries.summaries/xx_<cond1_id>_<cond2_id>.deltapsi.htmlfiles: interactive HTML5 summaries with MAJIQ quantifications.<cond1_id>_<cond2_id>.deltapsi.tsv: A tab-delimited file with all the genes and LSV quantifications and genomic information.